成池铉不良事件 【资料】不良事件和严重不良事件
不良事件和严重不良事件 1制定不良事件和严重不良事件的SOP 任何临床试验开始之前,申办者和研究者应制定关于不良事件的记录和严重不良事件报告的标准操作程序(SOP),应包括: 有关术语的定义,包括不良事件、严重不良事件、药物不良反应、非预期药物不良反应等。

不良事件的记录要求和快速报告的标准。 2不良事件和非预期药物不良反应 2.1不良事件(Adverse Event,AE) 不良事件是病人或临床试验的受试者接受一种药品后出现的不良医学事件,但不一定与治疗有因果关系。

按照GCP的要求,不管是否与试验用药有因果关系,研究者均应在原始记录中记录所有不良事件,并转抄至病例报告表中。不良事件也包括研究开始前存在的疾病发病次数和严重程度的增加。

不良事件的记录至少应包括: 不良事件的描述。 发生时间。 终止时间。 程度及发作频度。 是否需要治疗,如需要,请记录所给予的治疗。 研究者判断不良事件是否与应用试验药品有关。
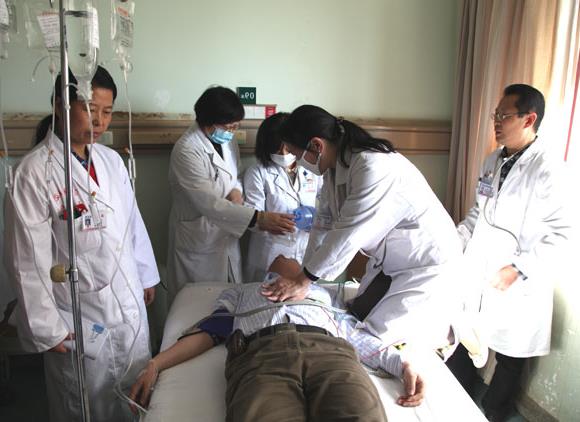
不良事件应追踪至解决。有关不良事件的医学文件均应记录在原始文件中,包括实验室检查的通知单(如:X线检查、心电图等)和检查结果报告单。如受试者因试验结束或受试者出院等而无法继续接受研究者的治疗,研究者应将受试者的病例摘要(包括治疗安排和不良事件是否需要继续随访的说明等)交给负责继续治疗他们的医生。
这些信息也要求记录在原始文件中。 2.2非预期药物不良反应(Unexpected Adverse the Reaction,UADR) 非预期药物不良反应是性质或严重程度与相应的试验药品资料不一致的药品不良反应。
动物试验和临床试验有很大的不同,很多临床前研究无法预见的医学事件会在临床试验中出现。
当多个受试者出现相同的不良事件,而在目前的研究者手册或研究方案中没有提到其性质、严重程度和频度与试验药品有关,那么研究者应尽快报告申办者这一不良事件。反复在不同的受试者身上出现相同的不良事件,对评价新药的安全性是非常重要的信息。
一个偶发的不良事件,研究者可能认为与试验药品无明显关系,当多个受试者出现或多个试验单位报告这一不良事件时就有可能成为事实上与试验药品有关的证据。 不良事件反复或多次发生,申办者或所委托的合同研究组织应和主要研究者一起研究有关信息,包括病史、以前的治疗、疾病的状况、合并用药及变化、使用试验药品的剂量和有无过量应用等。
如确认这一不良事件为非预期药物不良反应,应写出安全性报告交药品监督管理部门和伦理委员会,并通报所有参加同一药品试验(包括不同试验方案)的研究者,必要时应修改研究者手册,使其包括新的不良反应或已知不良反应的频度和严重程度的变化。
按照GCP进行新药临床试验与我国在安全性评价方法上有很大的不同。
以往的新药临床试验仅对药品不良反应记录、分析和写入总结报告。而按照GCP进行的临床试验,要求将临床试验中发生的所有不良事件均记录在病例报告表上,并将所有不良事件列表进行药物相关性分析。

对新药不良反应的报告和发现非预期药物不良反应是非常重要的步骤,使新药的安全性评价更科学可信。 3严重不良事件(Serious Adverse Event,SAE) 是临床试验过程中发生需住院治疗、延长住院时间、伤残、影响工作能力、危及生命或死亡、导致先天畸形等事件。


















{kind=link}